Opplæringsmateriell - veiledning for innsending og distribusjon
Publisert:
|
Oppdatert:
Endringer
- : Endret anbefaling av distribusjon av opplæringsmateriell for generiske legemidler
Når legemidler har spesielle risikoer for bivirkninger kan produsenten bli pålagt å lage materiell til opplæring av helsepersonell, pasienter og pårørende. Formålet med dette materiellet er å redusere risikoen for at disse bivirkningene skal oppstå.
Innhold på siden
Opplæringsmateriell er et risikominimeringstiltak (additional risk minimisation measures) og kan være et krav i forbindelse til markedsføringstillatelsen til legemidlet. Kravene til opplæringsmateriellet gis under behandlingen av søknaden om markedsføringstillatelsen (MT). For legemidler i sentral prosedyre gis kravene i Annex II, for øvrige prosedyrer beskrives de i risikohåndteringsplanen (RMP).
For å få oversikt over når MT-innehaver skal innsende opplæringsmateriell til DMP kan denne figuren brukes:
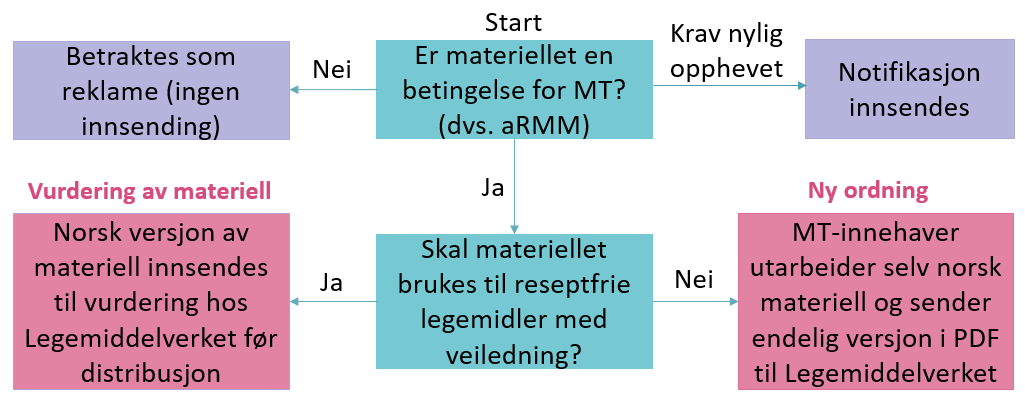
Innsendelse av opplæringsmateriell
Materiell til reseptfrie legemidler med veiledning
Når materiell skal brukes til reseptfrie legemidler med veiledning så skal materiellet vurderes og aksepteres av DMP innen det tas i bruk. Det betyr at alt materiell inkludert følgebrev, layout, målgrupper og distribusjonsform skal sendes til DMP for aksept før distribusjon:
Dette skal sendes inn:
-
opinion eller Kommisjonsvedtak (inkludert Annex II)
-
siste versjon av Risikohåndteringsplan (RMP)
-
opplæringsmateriell i word-format (bruk ”spor endringer” ved senere oppdateringer)
-
PDF-versjon av materiell med forslag til bildebruk og layout om det foreligger
-
dersom følgebrev skal være med utsendelsen skal også dette vurderes
-
forslag til mottakergrupper
Materiellet sendes til opplaeringsmateriell@dmp.no eller educationalmaterial@noma.no
Opplæringsmateriell i «ny ordning»
Fra 10. november 2020 skal MT-innehaver før distribusjon sende inn endelige norske versjoner av opplæringsmateriell som ikke skal brukes til reseptfrie legemidler med veiledning. DMP vurderer og aksepterer ikke materiellet før bruk. Materiellet blir arkivert internt og DMP fører stikkprøvebasert tilsyn med materiellet. Bakgrunn og evaluering av ordningen kan du finne her.
DMP har utviklet et onlinekurs (som du finner nederst på siden) som beskriver bakgrunnen for opplæringsmateriell og hva MT-innehaver skal være oppmerksom på under utformingen av opplæringsmateriell. Det finnes også en sjekkliste som fungerer som resyme av onlinekurset og en ordliste som kan brukes under arbeidet med opplæringsmateriell.
Innen bruk og distribusjon sendes inn følgende:
-
endelige norske PDF-versjoner av materiell med endelig layout (og evt. følgebrev)
-
nyeste versjon av risikohåndteringsplan del V
(for CP-preparater kananneks IID av SmPC sendes som alternativ)
-
mottakergrupper (MT-innehaver bestemmer mottakergrupper selv)
-
hvordan materiell skal distribueres inklusiv tidsplan for distribusjon
Ved oppdatering av materiell sendes i tillegg:
-
begrunnelse for oppdatering av materiell
Materiellet sendes til opplaeringsmateriell@dmp.no eller educationalmaterial@noma.no
Legemidler som allerede er markedsført, og får krav om opplæringsmateriell
For legemidler som allerede er markedsført, har MT-innehaver en frist på 30 dager på å sende inn opplæringsmateriell etter at kravet om opplæringsmateriell er godkjent.
Stikkprøvebasert tilsyn
DMP vil føre stikkprøvebasert tilsyn på materiell som ikke brukes til reseptfrie legemidler med veiledning. Tilsynet gjøres for å sikre at opplæringsmateriell er i tråd med anbefalinger fra EMA og for å sikre at opplæringsmateriell oppfyller individuelle krav til hvert opplæringsmateriell. Dersom DMP finner avvik fra EMAs anbefalinger eller individuelle krav til hvert legemiddel, tar vi kontakt til MT-innehaver.
Veiledninger
Veiledning for utarbeidelse av opplæringsmateriell:
Guideline on good pharmacovigilance practices (GVP) Modul XVI Addendum I – Educational materials
Overordnet beskrivelse av ulike risikominimeringstiltak og opplæringsmateriell: Guideline on good pharmacovigilance practices (GVP) Module XVI– Risk minimisation measures: selection of tools and effectiveness indicators
Sjekkliste for utforming av opplæringsmateriell
Språk og tekst
Opplæringsmateriellet skal være skrevet på tydelig og forståelig norsk, og helst uten tunge og kompliserte setninger. Hvis språket er av dårlig kvalitet eller uforståelig vil firmaet bli bedt om å forbedre språket ved en vurdering/stikkprøvebasert tilsyn. Språklig korrektur av pasientmateriell prioriteres som utgangspunkt høyere enn materiell til helsepersonell.
Som tommelfingerregel skal det hverken stå mer eller mindre enn punktene som er beskrevet i RMP del V/Annex IID (punktliste for hva aRMM skal inneholde). All unødig tekst i opplæringsmateriell som ikke er beskrevet i RMP del V/annex IID bør generelt slettes siden hensikten med opplæringsmateriell er å beskrive utvalgte risikoer ved bruk av legemidlet.
Eksempler på dette kan være virkningsmekanismer, sykdomslære osv. noen ganger beskrevet i opplæringsmateriell. Dette skal bare bli stående hvis det spesifikt er beskrevet i RMP/Annex IID.
Farge-, bilde- og logobruk
Opplæringsmateriell skal ikke inneholde farger som er kommersielt relatert, dvs. farger som er relatert til markedsføring av produktet. Det er imidlertid lov å bruke firma sine farger i materiellet.
Firma har lov å bruke firmalogo ét sted på hvert dokument (eks. på enten for- eller bakside).
Opplæringsmateriellet skal ikke inneholder bilder eller illustrasjoner med mindre dette bidrar til økt forståelse av innholdet, f.eks. en bestemt prosedyre, ulike injeksjonssteder/teknikker for administrering eller lignende.
Fargebruk må ikke bidra til dårlig lesbarhet ved redusert kontrast.
Signatur fra forskriver/pasient
DMP ønsker ikke at opplæringsmateriell inneholder plass/krav til signatur fra forskriver og/eller pasient med mindre dette er vedtatt av EMA/Kommisjonen. En slik signatur kan gi feilaktig inntrykk av at det følger med ekstra forpliktelser for pasient eller helsepersonell som signerer. Dette er ikke til hinder for at kontaktopplysninger til behandlende lege/klinikk og annet kan påføres pasientmaterialet når hensikten er å informere annet helsepersonell om kontaktopplysninger ved en krisesituasjon for pasienten.
Versjonssnummer
Firma må ha en form for versjonsnummer på hver side av opplæringsmateriellet for å muliggjøre oversikten over ulike utgaver. Firma skal sette på dette, hvis det mangler.
Følgebrev
Det anbefales at firma legge ved et følgebrev som forklarer hensikten med, evt. endringene i opplæringsmateriellet hvis materiellet skal distribueres til mottaker og ikke kun publiseres på nett. Firma bør oppfordres til å merke følgebrevet med vår standardlogo for sikkerhetsinformasjon.
Følgebrevet skal dessuten alltid inneholde opplysninger om hvor opplæringsmateriellet kan finnes f.eks. på Felleskatalogen.no:
Se oppdatert preparatomtale (SPC) og opplæringsmateriell på www.felleskatalogen.no.
Sikkerhetsinformasjonslogo
DMPs sikkerhetsinformasjonslogo for opplæringsmateriell er et krav og skal være på materiell, følgebrev og konvolutter, for å blant annet hjelpe helsepersonell og pasienter å skille opplæringsmateriell og reklamemateriell. På pasientkort kan firma vurdere om det er plass til logoen.
Vær oppmerksom på at logoen er annerledes enn sikkerhetsinformasjonslogoen for Kjære helsepersonell-brev, som tidligere er brukt på opplæringsmateriell.
Standardtekster
Melding av bivirkninger
I følgebrev og opplæringsmateriell hvor det oppfordres til å melde bivirkninger kan følgende standardsetninger brukes:
Tekst til materiell til helsepersonell
-
Helsepersonell bes melde nye, uventede og alvorlige mistenkte bivirkninger på elektronisk meldeskjema:
Tekst til materiell til pasienter
-
Bivirkninger kan meldes på elektronisk skjema til DMP:
For legemidler med svart trekant vil standardtekst om å melde bivirkninger komme etter svart trekant og forklarende tekst om særlig overvåking. Se avsnitt om svart trekant nedenfor.
Unntak er pasientkort hvor det er ønskelig å ikke inkludere svart trekant pga. sparsomt med plass. Her kan følgende standardsetning om å melde bivirkninger brukes:
- Du kan bidra ved å melde enhver mistenkt bivirkning, se www.dmp.no/pasientmelding
Standardsetning til hvor materiell kan finnes på nett
Det bør fremgå av opplæringsmateriell og følgebrev hvis det er aktuelt, hvor opplæringsmateriellet og preparatomtalen kan finnes på nettet. Følgende standardsetning kan brukes:
Se oppdatert preparatomtale (SPC) og opplæringsmateriell på www.felleskatalogen.no.
Svart trekant
Opplæringsmateriell for legemidler som står oppført på felles europeisk overvåkingsliste skal inneholde svart trekant ét sted på alle dokumenter (unntak av pasientkort*) med forklarende tekst. Den svarte trekanten bør plasseres foran preparatnavnet (▼preparatnavn) i begynnelsen av opplæringsmateriellet, og ha samme høyde som bokstavene i preparatnavnet den står foran. På samme side bør det være en forklarende tekst til den svarte trekanten. Forklarende tekst til materiell til:
Helsepersonell:
-
Dette legemidlet er underlagt særlig overvåking for å oppdage ny sikkerhetsinformasjon så raskt som mulig. Helsepersonell oppfordres til å melde enhver mistenkt bivirkning på elektronisk meldeskjema:
Pasienter:
-
Dette legemidlet er underlagt særlig overvåking for å oppdage ny sikkerhetsinformasjon så raskt som mulig. Du kan bidra ved å melde enhver mistenkt bivirkning, se
*EMA ønsker at svart trekant inklusiv forklarende tekst ikke er inkludert i pasientkort siden det ofte er sparsom med plass på pasientkort. Plassen skal derfor spares til å øke lesbarhet av annen informasjon på pasientkortet. Oppfordring til å melde bivirkninger skal derimot være på pasientkort også.
Distribusjon
Firma skal alltid sende inn forslag til om materiell skal distribueres og/eller publiseres på FK. Firma skal i tillegg foreslå tidsplan for publisering og/eller distribusjon. Opplæringsmateriale skal vanligvis distribueres til utvalgte mottakergrupper ved nytt opplæringsmateriale eller ved betydningsfulle endringer i eksisterende opplæringsmateriell. Ved mindre endringer kan det vurderes å kun publisere på FK.
Materiell for generiske legemidler trenger som utgangspunkt ikke å bli distribuert. Originalpreparatet har typisk tidligere distribuert opplæringsmateriellet, men MT-innehavere må sikre seg at helsepersonell er klar over at det finnes opplæringsmateriell for sitt produkt, og at materiellet kan bestilles fysisk fra MT-innehaver, f.eks. via e-post.
Apotek
Opplæringsmateriell til apotek sendes på epost. Oversikt over epostadresser til apotekene fås fra Felleskatalogen. For å få utlevert listene sendes en epost til redaksjonen@felleskatalogen.no med emnefeltet "E-post apotek til utsendelse av opplæringsmateriell".
Eposten til apoteket bør inneholde teksten "Sikkerhetsinformasjon etter krav fra DMP" i emnefeltet. Opplæringsmateriellet legges ved eposten i pdf format.
Fastleger/allmennleger
Opplæringsmateriell skal ikke skal sendes til fastleger på papir. DMP formidler isteden informasjonen via varsler i pasientjournalen (EPJ).
Det varsles utelukkende om oppdatert materiell hvis oppdateringen er krevd av myndigheter. Det er derfor viktig at det tydelig fremgår i innsendelse til DMP om oppdateringen er krevd av myndighetene. Det må i tillegg tydelig fremgår at fastleger er mottakergruppe for materiellet.
Øvrige leger/annet helsepersonell
Opplæringsmateriell til øvrige grupper av helsepersonell sendes per post.
Publisering hos Felleskatalogen
Vi oppfordrer på det sterkeste til at alt opplæringsmateriell publiseres på Felleskatalogen. Grunnen til dette er at Felleskatalogen er brukt av stort sett all helsepersonell og det er en større sannsynlighet for at opplæringsmateriale blir brukt hvis det er lett tilgjengelig. Det vil derfor øke pasientsikkerheten hvis all opplæringsmateriell er tilgjengelig for helsepersonell og pasienter via Felleskatalogen.
Det finnes tre kategorier for å legge opp opplæringsmateriell på FK:
- Opplæringsmateriell og veiledning ved bruk
- Graviditetsforebyggende program
- Opplæringsmateriell og graviditetsforebyggende program
Felleskatalogen sorterer materiell i korrekt kategori når materiell er innsendt til Felleskatalogen.
- Opplæringsmateriell (RMP-materiell) publiseres på www.felleskatalogen.no
- Bruk FK SHARE for opplasting, https://share.felleskatalogen.no/
- Er du ny FK SHARE-bruker? Send e-post til redaksjonen@felleskatalogen.no for å bli registrert
NB! RMP-materiell skal alltid merkes med ‘RMP’ i filnavnet. RMP-materiell er etter krav fra DMP, og Felleskatalogens redaksjon vil ikke vurdere dette materiellet ytterligere.
Opplæringsmateriell for generiske/biotilsvarende legemidler
Der betingelsene for generiske/biotilsvarende produkter er identiske med originalproduktet, bør opplæringsmateriellet harmoniseres og følge det eksisterende godkjente materialet for originalproduktet (GVP Module XVI Addendum 1.2.)
Felles utarbeidelse og distribusjon
MT-innehavere for ulike produkter med samme virkestoff og identiske vilkår oppfordres til å samarbeide om utarbeidelsen av opplæringsmateriellet. MT-innehaverne bør kun sende inn ett forslag til opplæringsmateriell til DMP. Vi anbefaler også at MT-innehaverne samarbeider om distribusjon av opplæringsmateriellet for å sikre at mottakerne på distribusjonslisten får samme felles materiell.
Onlinekurs
Modul 1: Generelt om legemiddelovervåking og opplæringsmateriell (youtube)
Modul 2: Språk, budskap og målgruppe (youtube)
Modul 3: Innsendelse til DMP og viktige ting å huske på (youtube)