Søknad om klinisk utprøving
Publisert:
|
Oppdatert:
Endringer
Informasjon om saksbehandling, gebyrer, regler for offentliggjøring og konfidensialitet.
Innhold på siden
Før søknaden kan opprettes
Clinical Trials Information System (CTIS) er en felles nettportal for innsending av søknader og samhandling om kliniske utprøvinger i EU og EØS.
Man må ha en EMA-konto for å få tilgang til CTIS før innsending av en ny søknad om klinisk utprøving. Vennligst se vår nettside om hvordan man kan få tilgang til CTIS, saksbehandling av søknader i CTIS, informasjon om nettbasterte opplæring og nyttige opplæringsmateriell.

Veiledning og råd til bedrifter og akademia
DMP bistår bedrifter og akademia med veiledning og råd innenfor utvikling av medisinske produkter, klinisk utprøving og helseøkonomi. DMP veileder også apotek, grossister og tilvirkere på legemiddelområdet.
Les mer
Søknad om ny klinisk utprøving
En søknad om klinisk utprøving består av to deler. Del I består av dokumentasjonen som i en multinasjonal studie er felles for alle deltakende medlemsland, blant annet protokoll, IB og IMPD. Del II inneholder dokumentasjon som er tilpasset hvert enkelte medlemsland, blant annet pasientinformasjon, samtykkeskjema og forsikringsbevis.
Dersom Norge er foreslått og har sagt seg villig til å være den rapporterende medlemsstaten (RMS) vil vi lede og koordinere valideringen og utredningen av fellesdokumentasjon for del I.
Sponsoren må alltid sende inn del I av søknaden, eller velge å sende inn del II til ett eller flere medlemsland samtidig med del I. Alternativt kan sponsor sende inn del II til de ulike medlemslandene i etterkant, men senest to år etter at del I er godkjent. Sponsor skal da bekrefte at det ikke er lagt til ny vesentlig vitenskapelig informasjon som gjelder del I. Dersom sponsor ikke sender inn del II innen to år etter konklusjonen for del I, bortfaller søknaden. En søknad om vesentlig endring kan ikke sendes før både del I og del II er innsendt og søknaden godkjent i alle medlemsland.
For kliniske utprøvinger som kun går i Norge (mononasjonale søknader), må del I og II sendes inn samtidig.
Mer informasjon om:
Saksbehandlingstid for søknad om klinisk utprøving
For søknader om kliniske utprøvinger, definerer forordningen (CTR) frister for sponsor og myndigheter. Disse er oppsummert i figuren nedenfor.
Ved innsending av søknad i CTIS, får man tilgang til oversikten over tidslinjer og frister under «timetable». Denne er til enhver tid gjeldende for søknaden.
Fristene er basert på kalenderdager, men nasjonale helligdager gjør at noen frister forlenges. I tillegg vil enkelte frister aldri falle på en mandag. Etterfølgende frister endres dersom sponsor eller myndigheter leverer tidligere enn den frist som er gitt i CTIS.
Det er ikke teknisk mulig å forlenge fristene. Dersom frister ikke overholdes vil søknaden forfalle («lapse»). Det er ikke mulig å gjenåpne en forfalt søknad, denne må innsendes på nytt.
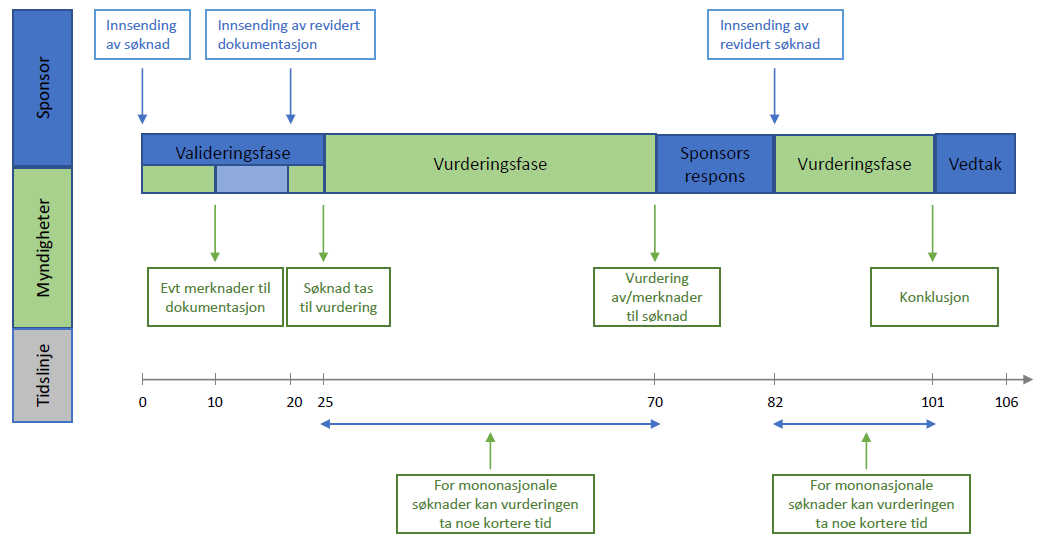
Saksbehandling av en søknad om klinisk utprøving er inndelt i 3 faser:
Valideringsfasen
-
10 dager (ved spørsmål til søker kan det legges til 15 dager – 25 dager til sammen)
-
I denne perioden kan RMS be om tilleggsdokumentasjon slik at søknaden er komplett før utredningen starter. Sponsor får vanligvis maksimalt 10 dager til å svare på eventuelle spørsmål.
-
Sponsor vil motta en beslutning på om søknaden er valid innen 5 dager etter at alle spørsmål er besvart.
Vurderingsfasen
-
45 dager fra valideringskonklusjon. Ved spørsmål til søker kan det legges til 31 dager – 76 dager til sammen.
-
Dersom søknaden involverer utprøving av legemiddel som er definert som avansert terapi (ATMP) kan RMS forlenge vurderingsfasen med maksimalt 50 dager.
-
Dersom myndighetene har spørsmål til søknaden (RFI), vil dette forlenger vurderingsfasen med maksimalt 31 dager - 12 dager for sponsor til å svare på spørsmål og ytterligere 19 dager for vurdering av sponsors respons.
-
Sponsor vil motta konklusjon på Del I av søknaden i CTIS.
Beslutningsfasen (Decision)
-
Sponsor vil motta endelig avgjørelse innen 5 dager etter at både Del I og Del II har blitt konkludert.
-
Søknaden om klinisk utprøving kan bli godkjent, godkjent med vilkår eller avslått.
Regler for publisering av personopplysninger og kommersielt konfidensiell informasjon
All informasjon i CTIS vil i utgangspunktet gjøres offentlig, men noe informasjon kan holdes tilbake. Dette gjelder:
-
Personopplysninger
-
Kommersielt konfidensiell informasjon
-
Konfidensiell kommunikasjon mellom medlemsstatene (i forbindelse med at en søknad utredes)
-
Informasjon knyttet til tilsyn med kliniske utprøvinger
Det er sponsors ansvar å sikre samsvar med forordning (EU) nr. 2016/679 og forordning (EU) nr. 2018/1725 ved opplasting av dokumenter og behandling av personopplysninger i CTIS.
Ved innsendelse av dokumenter som trenger skjerming (for eksempel personopplysninger, signaturer el.), skal det lastes opp to versjoner av dokumentet i CTIS;
-
en versjon der relevant informasjon er sladdet og markert med
«for publication» , og
-
en versjon som ikke er sladdet og markert med «not for publication».
-
Den første versjonen som lastes opp blir markert «for publication».
Regler for tidspunktet for offentliggjøring
Tidspunktet for offentliggjøring av dokumentasjon styres av hvilken kategori den kliniske utprøvingen er i:
-
Category 1 trials - Pharmaceutical development
-
Category 2 trials- Therapeutic exploratory and confirmatory clinical trials
-
Category 3 trials - Therapeutic use clinical trials
Det er sponsor som definerer hvilken kategori studien tilhører ved innsendelse av søknad. Se «Appendix on transparency rules - EMA» for mer informasjon om regler for offentliggjøring av dokumentasjon knyttet til en klinisk utprøving.
Gebyrer
Gebyrer for søknader om klinisk utprøving av legemidler til mennesker gjeldende fra 01.01.2023
| Type søknad | Satser |
|---|---|
| Ny søknad – Norge som referanseland (Forordning nr. 536/2014) | 70 000 NOK |
| Ny søknad – Norge som berørt land (Forordning nr. 536/2014) | 30 000 NOK |
| Endringssøknad (Direktiv EC 2001/20 og Forordning nr. 536/2014) | 6 000 NOK |
| Søknad om overføring til Forordning nr. 536/2014 | Ingen gebyr |
Det vil ikke bli krevd gebyr for ikke-kommersielle studier.
Fakturainformasjon må lastes opp under “Proof of Payment of fee" under “Form", del I av søknaden i CTIS. Det vil bli sendt en faktura etter at søknaden er mottatt. Fakturaen vil påføres EU CT nummer og DMPs saksnummer.
Krav til språk
Søknaden må skrives på engelsk. Protokollsynopsis og informasjon som dekkes av punkt 24, 59–73, Annex I til forordningen (CTR) skal være skrevet på norsk. Annen dokumentasjon som er rettet mot forsøkspersoner (pasientinformasjon, samtykkeskjema, osv.) skal også skrives på norsk.