Krav til produsenter
Publisert:
|
Oppdatert:
Endringer
Produsent er en fysisk eller juridisk person som framstiller eller helrenoverer utstyr, eller som får utstyr designet, framstilt eller helrenovert, og som markedsfører nevnte utstyr i eget navn eller under eget varemerke.
Innhold på siden
Hvilke krav gjelder en produsent
I tabellen nedenfor er noen av kravene listet opp, men listen er ikke fullstendig. Hvilke krav som er relevante for hver enkelt produsent avhenger også av hvilken type medisinsk utstyr det er og utstyrets risikoklassifisering. Krav og forpliktelser til produsenter finnes flere steder i MDR og IVDR og det er viktig at produsenter setter seg inn i hele regelverket.
| Artikkel | Oversikt over noen av kravene |
|---|---|
| MDR Artikkel 10 IVDR Artikkel 10 |
Generelle krav til:
Se egen side for spesielle krav til individuelt tilpasset utstyr. |
|
MDR Artikkel 11 IVDR Artikkel 11 |
Krav om autorisert representant for produsenter utenfor EU/EØS-områdetog Tyrkia |
|
MDR Artikkel 15 IVDR Artikkel 15 |
Krav om person med ansvar for overholdelse av regelverket |
| MDR Artikkel 18 |
Krav om pasientinformasjon og implantatkort for implanterbart medisinsk utstyr |
| MDR Artikkel 22 | Krav til system og prosedyresett |
|
MDR Artikkel 29 IVDR Artikkel 26 |
Krav til registrering av utstyr |
|
MDR Artikkel 31 IVDR Artikkel 28 |
Krav til registrering av produsenter og individuelt registreringsnummer |
|
MDR Artikkel 32 IVDR Artikkel 29 |
Krav til sammendrag om sikkerhet og klinisk ytelse (MDR)/ytelse (IVDR) |
Oversikt over de ulike rollene for markedsdeltakere
| Markedsdeltaker | Definisjon i MDR artikkel 2 og IVDR artikkel 2 |
|---|---|
| Produsent | en fysisk eller juridisk person som framstiller eller helrenoverer utstyr, eller som får utstyr designet, framstilt eller helrenovert, og som markedsfører nevnte utstyr i eget navn eller under eget varemerke. |
| Autorisert representant | enhver fysisk eller juridisk person etablert i Unionen* som har fått og har akseptert en skriftlig fullmakt fra en produsent plassert utenfor Unionen, til å utføre bestemte oppgaver på dennes vegne med hensyn til dennes forpliktelser i henhold til denne forordning. |
| Importør | enhver fysisk eller juridisk person etablert i Unionen* som bringer utstyr fra en tredjestat i omsetning i Unionen*. |
| Distributør | enhver fysisk eller juridisk person i omsetningskjeden, utenom produsenten eller importøren, som gjør utstyr tilgjengelig på markedet fram til ibruktaking. |
| Helseinstitusjon | en organisasjon som har som hovedformål å pleie eller behandle pasienter eller fremme folkehelsen. |
* Med «Unionen» menes her EU/EØS-området og Tyrkia.
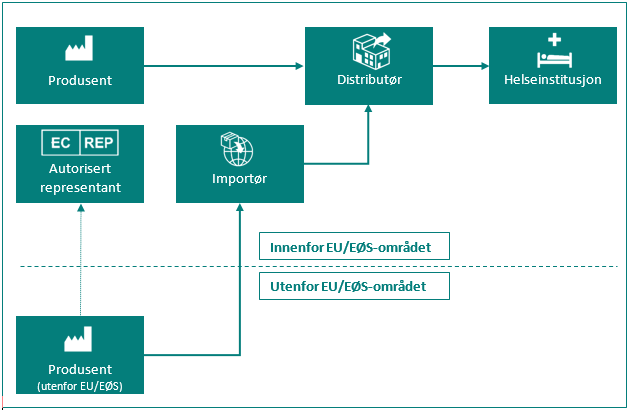
Figur: Markedsdeltaker i omsetningskjeden.